Discriminating somatic and germline mutations in tumor DNA samples without matching normals
July 2015
Genome Research
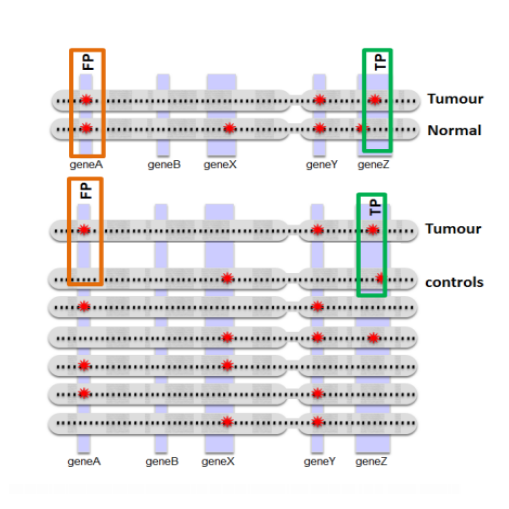
Tumor analyses commonly employ a correction with a matched normal (MN), a sample from healthy tissue of the same individual, in order to distinguish germline mutations from somatic mutations. Since the majority of variants found in an individual are thought to be common within the population, we constructed a set of 931 samples from healthy, unrelated individuals, originating from two different sequencing platforms, to serve as a virtual normal (VN) in the absence of such an associated normal sample.
READ MORE
FuMa: reporting overlap in RNA-seq detected fusion genes
December 2015
Bioinformatics
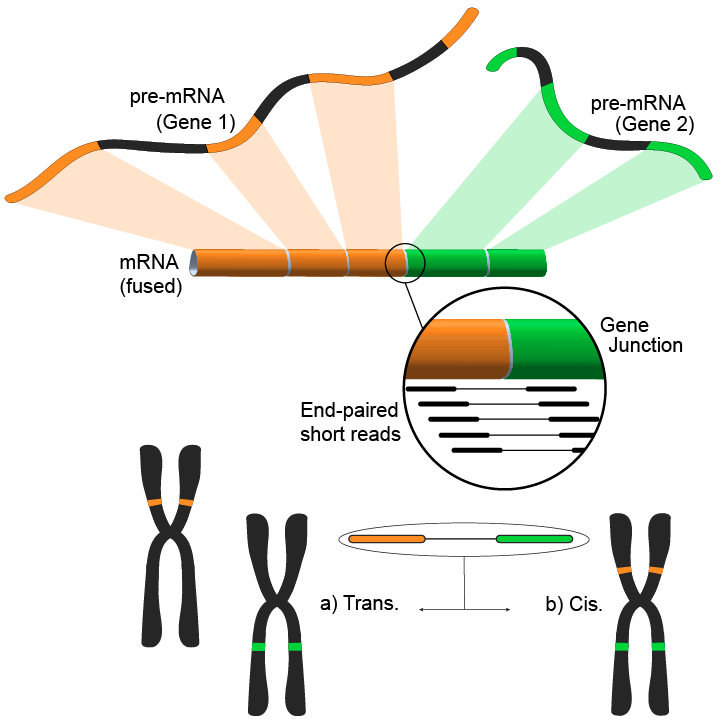
A new generation of tools that identify fusion genes in RNA-seq data is limited in either sensitivity and or specificity. To allow further downstream analysis and to estimate performance, predicted fusion genes from different tools have to be compared. However, the transcriptomic con-text complicates genomic location-based matching. FusionMatcher (FuMa) is a program that reports identical fusion genes based on gene-name annotations.
READ MORE
ImmunoGlobulin galaxy (IGGalaxy) for simple determination and quantitation of immunoglobulin heavy chain rearrangements from NGS
December 2014
BMC Immunology
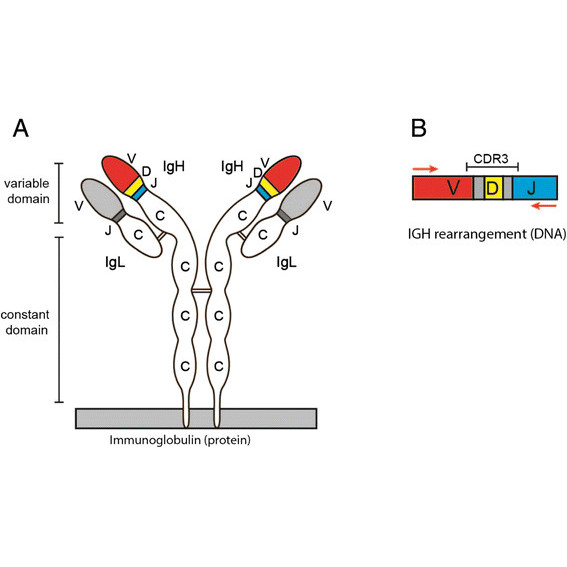
In this study we describe ImmunoGlobulin Galaxy (IGGalaxy)- a convenient web based application for analyzing next-generation sequencing results and reporting IGH gene rearrangements for both repertoire and clonality studies.
READ MORE
Integration of EGA secure data access into Galaxy
December 2016
F1000 Research
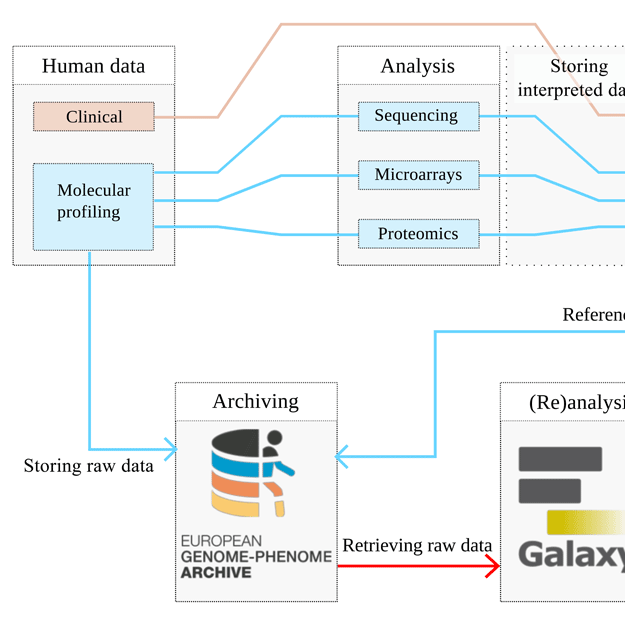
High-throughput molecular profiling techniques are routinely generating vast amounts of data for translational medicine studies. Secure access controlled systems are needed to manage, store, transfer and distribute these data due to its personally identifiable nature. The European Genome-phenome Archive (EGA) was created to facilitate access and management to long-term archival of bio-molecular data.
READ MORE
iReport: a generalised Galaxy solution for integrated experimental reporting
October 2014
GigaScience
Galaxy offers a number of visualisation options with components, such as Trackster, Circster and Galaxy Charts, but currently lacks the ability to easily combine outputs from different tools into a single view or report. We have developed a generic and flexible reporting tool for Galaxy, iReport, that allows users to create interactive HTML reports directly from the Galaxy UI, with the ability to combine an arbitrary number of outputs from any number of different tools.
READ MORE
NanoGalaxy: Nanopore long-read sequencing data analysis in Galaxy
October 2020
GigaScience
NanoGalaxy has been developed for the analysis of nanopore data and is suitable for diverse applications, including de novo genome assembly from genomic, metagenomic, and plasmid sequence reads.
READ MORE